Different types of European legislation
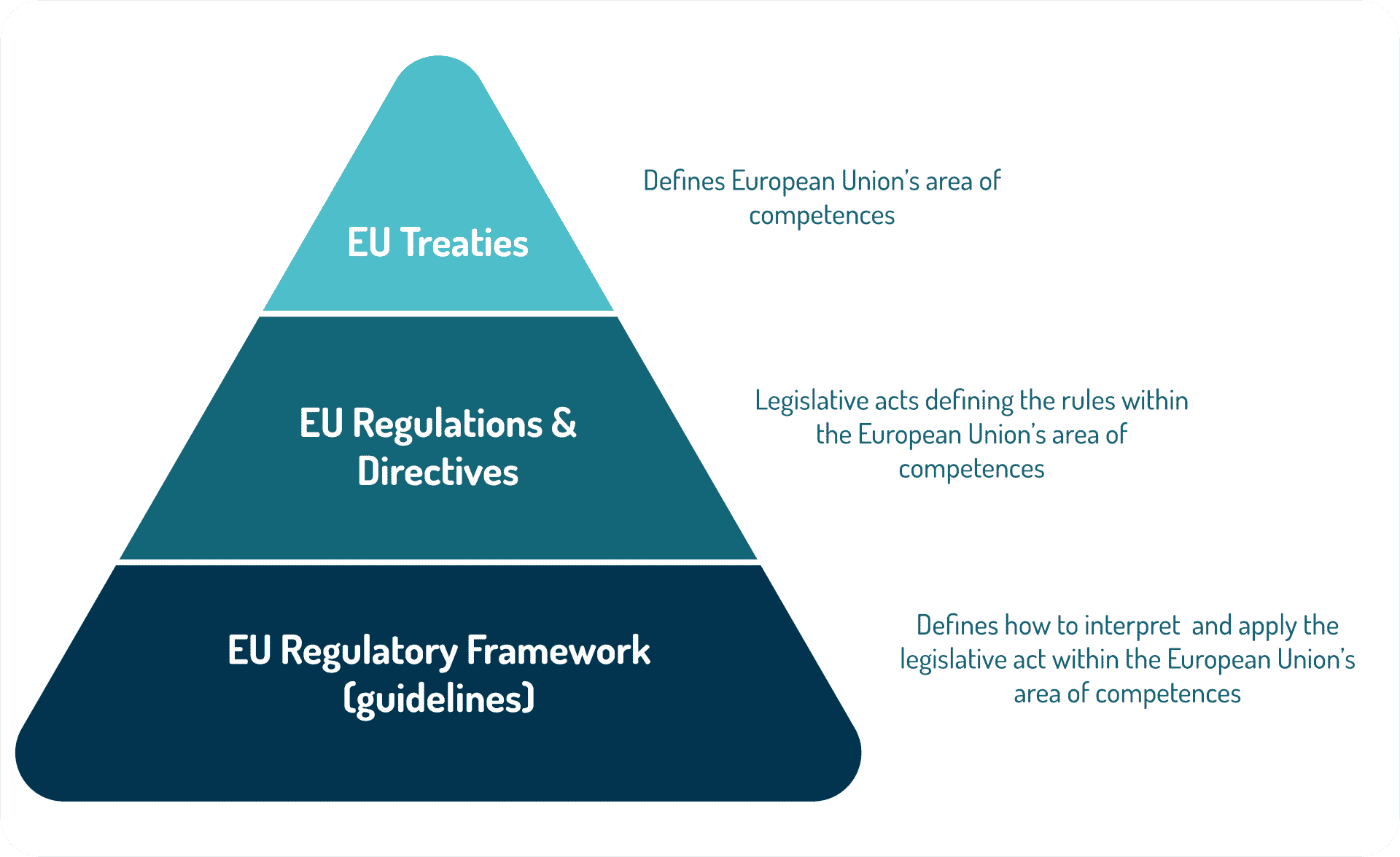
The European Union (EU) is based on the rule of law. This means that every action taken by the EU is founded on treaties that have been approved voluntarily and democratically by all EU member countries. For example, if a policy area is not cited in a treaty, the European Commission cannot propose a law in that area.
A treaty is a binding agreement between EU member countries. It sets out EU objectives, rules for EU institutions, how decisions are made and the relationship between the EU and its member countries.
Two treaties form the constitutional basis of the European Union (EU):
- The Treaty on European Union (TEU; also referred to as the Treaty of Maastricht), establishing the European Union, its governance and its central institutions (such as the European Commission, European Parliament, and Council of the European Union)
- The Treaty on the Functioning of the European Union (TFEU; also referred to as the Treaty of Rome) establishing the European Economic Community and creating a single market for goods, labour, services, and capital across Member states.
The aims set out in the EU treaties are achieved by several types of legal or legislative acts (EU legislation).

A “regulation” is a binding legislative act.
Regulations are binding in their entirety for all EU countries. They apply automatically and uniformly to all EU countries as soon as they enter into force, without needing to be transposed into national law.
A “directive” is a legislative act that sets out a goal that all EU countries must achieve.
Directives require EU countries to achieve a certain result, but leave them free to choose how to do so. EU Member states must transpose directives into national law in order to realise the objectives set out by the directive. Transposition into national law must take place by the deadline set when the directive is adopted (generally within 2 years).
EMA Scientific Guidelines
EMA committees prepare scientific guidelines in consultation with regulatory authorities in the European Union (EU) Member States to help applicants prepare marketing authorisation applications for human or veterinary medicines. Guidelines reflect a harmonised approach of the EU Member States and EMA on how to interpret and apply the requirements for the demonstration of quality, safety and efficacy set out in the Community directives.
EMA strongly encourages applicants and marketing authorisation holders to follow these guidelines. Applicants need to fully justify deviations from guidelines in their applications at the time of submission. Before that, they should seek scientific advice, to discuss any proposed deviations during medicinal product development.
The topics covered by these scientific guidelines are related to the different phases of development, the quality controls and specific types of products (such as vaccines, plasma-derived medicinal products, ATMP,…).
ICH Guidelines
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) is unique in bringing together the regulatory authorities and pharmaceutical industry to discuss scientific and technical aspects of pharmaceuticals and develop ICH guidelines. ICH’s mission is to achieve greater harmonisation worldwide to ensure that safe, effective and high-quality medicines are developed, and registered and maintained in the most resource-efficient manner whilst meeting high standards. ICH now includes 19 Members and 35 Observers.
ICH guidelines are categorised along 4 broad workstreams:
- Quality guidelines (the “Q-family”): manufacture, control and stability of ingredients and products
- Efficacy (the “E-family”): design, conduct, safety and reporting of clinical trials
- Safety (the “S-family”): Non-clinical test strategies and methods
- Multidisciplinary (the “M-family”): diverse cross-cutting topics, including CTD/eCTD, medical dictionary and e-standards for transfer of regulatory information.
In addition, ICH has developed an extensive set of Q&A documents and training materials to further clarify concepts and principles in ICH Guidelines.
European Pharmacopoeia monographs and chapters
European Pharmacopoeia monographs and chapters and EMA scientific guidelines are complementary instruments to ensure the quality of medicinal products. The European Pharmacopoeia is a collection of standardised specifications on the quality of pharmaceutical preparations, their constituents or their containers.
Monographs and general chapters have legal force with regard to the quality part of the dossiers supporting marketing authorisations unless the scope of the particular monograph or general chapter makes it clear that the text is not mandatory.
